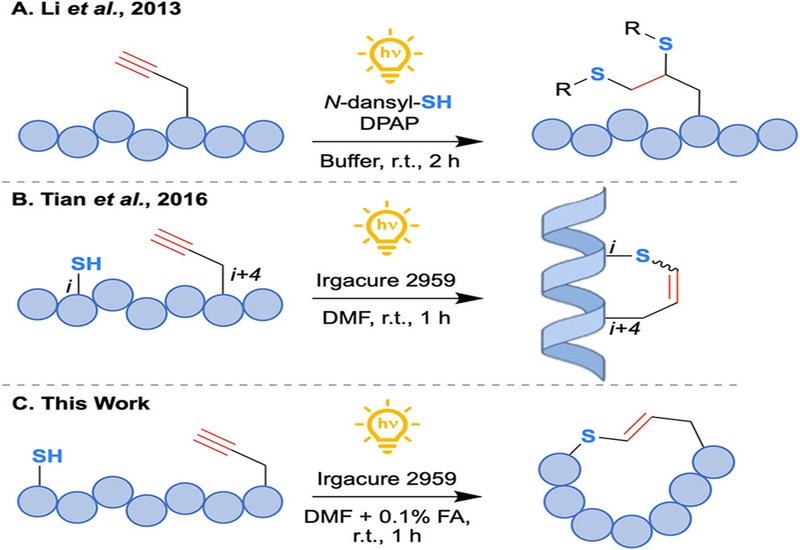
Disulfide bridges are nature's preferred way to cyclize neuropeptides such as oxytocin and somatostatin, but the bond is a pharmacological liability: reducing agents present at high concentrations inside cells cleave it rapidly, shortening plasma half-life and limiting therapeutic potential. Chemists have long sought bioisosteres that mimic the disulfide geometry while surviving the intracellular redox environment. Triazole formation and ring-closing metathesis each solve part of the problem, yet both typically demand metal catalysts that are difficult to remove and require the synthesis of unnatural amino acid building blocks. A metal-free, mild alternative compatible with fully unprotected peptides has remained elusive.
Researchers in the Scanlan Group at Trinity College Dublin and the Muttenthaler Group at the University of Vienna, published in the Journal of the American Chemical Society, developed a photochemical radical thiol–yne macrocyclization strategy that converts linear, cysteine-containing peptides into vinyl sulfide-bridged macrocycles under UV-A or blue-LED irradiation. The team optimized conditions around Irgacure 2959 as the photoinitiator in DMF with 0.1% formic acid at 1.5 mM peptide concentration, achieving conversions above 95% in one hour without stirring. A complementary aqueous protocol using thioxanthen-9-one as a visible-light photosensitizer in 1:1 water/acetonitrile provided 84% conversion, broadening compatibility with biologically sensitive substrates.
The method delivered separable (E)- and (Z)-vinyl sulfide isomers for a series of oxytocin and carbetocin analogues spanning ring sizes up to 39-membered macrocycles, and extended to somatostatin and the approved melanocortin drug setmelanotide. A robustness screen modeled on the Collins–Glorius protocol confirmed tolerance to thiols, alkenes, alkynes, aldehydes, amines, halides, and all 20 canonical amino acids, with no meaningful inhibition of cyclization observed in any case. Conformational analysis by computational modeling revealed that the (Z)-oxytocin analogue 6Z is dominated by a single conformer accounting for 99.8% of the conformational space, closely paralleling endogenous oxytocin, whereas the (E)-isomer 6E distributes across eight conformers with populations ranging from 1% to 35%.
Functional IP1 assays on HEK-293 cells stably overexpressing the human oxytocin receptor showed that 6Z carries an EC50 of 3.22 nM, a twofold improvement over oxytocin itself (EC50: 6.65 nM), while 6E is approximately tenfold less potent (EC50: 55.67 nM). Neither analogue showed significant activity at the vasopressin receptors hV1aR or hV1bR, indicating receptor selectivity. For the setmelanotide series, the (Z)-configured analogue 21Z retained subnanomolar potency at the human melanocortin 4 receptor (EC50: 0.29 nM) compared to the parent disulfide peptide (EC50: 0.13 nM). Glutathione stability assays at 1,000-fold excess demonstrated that oxytocin and setmelanotide are completely reduced within five minutes, whereas all four vinyl sulfide analogues remained fully intact across the four-hour experiment. The team also demonstrated an on-resin thiol–yne cyclization followed by ionic addition of 1,2-ethanedithiol during TFA cleavage, furnishing a free-thiol handle for subsequent maleimide conjugation to install a fluorophore or biotin tag without additional chromatographic purification.
The thiol–yne macrocyclization platform addresses three longstanding challenges simultaneously: it operates metal-free under mild photochemical conditions, it generates a redox-stable vinyl sulfide that more closely mirrors disulfide geometry than triazole or alkene staples, and it provides direct access to both vinyl sulfide stereoisomers as separable compounds for structure–activity exploration. The (Z)-oxytocin analogue 6Z emerges as a lead compound for developing selective oxytocin receptor agonists, and the sequential cyclization-tagging protocol opens a concise route to labeled peptide probes for cell imaging and affinity-based pull-down studies. Taken together, the approach provides a broadly applicable tool for peptide drug development and library synthesis.